CAUTION: Service is experimental.
Refinement protocol
To reduce the discrepancy between the model structure factors and the observed data, while maintaining good geometry, we minimize the following target function: \(T_{total} = T_{Amber} + w_{xray}T_{xray}\).
- \(T_{Amber}\) represents Amber force field potential, specifically, we use ff14SB parameters.
- As the crystallogaphic component, we use maximum likelihood target \(T_{xray}\) in the form of negative logarithm of the likelihood.
- Throughout the refinement we modify the relative weight \(w_{xray}\) of the target components in the manner described below.
To make use of the high quality physics-based force field, we employ explicit interstitial solvent and explicit unit cell with periodic boundary conditions. In our protocol the following steps occur:
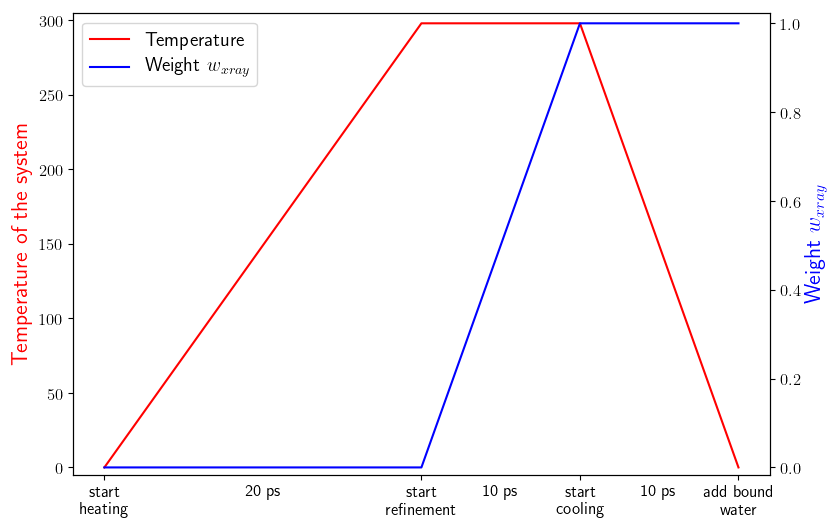
- After the system containing the unit cell is prepared (e.g. macromolecules are solvated, etc.), it is being heated up to the room temperature in 20 ps. This step is being affected only by the \(T_{Amber}\) term of the target potential.
- Next, while the system maintains the constant temperature, we gradually introduce the crystallographic restraints \(T_{xray}\) for 10 ps.
- For another 10 ps, we keep the crystallographic restraints \(T_{xray}\) constant. During this period, we gradually cool down the system.
- Finally, we clean up the system from the solvent which was used to model non-bonded interactions, and add crystallographic bound water molecules based on the electron density map peaks.
Schematic representation of the basic protocol: